FAQ's zur MDR Zertifizierung
MDR Conformity Assessment
The Medical Device Regulation (MDR, Regulation (EU) 2017/745) has been fully in force since May 2021 and replaces the previous Medical Devices Directive 93/42/EEC (MDD). As an EU regulation, it applies directly in all member states and imposes increased requirements on the safety, performance and traceability of medical devices. The MDR defines comprehensive regulations for the manufacture, distribution and monitoring of medical devices with the aim of improving patient safety.
[11.08.2025]
Conformity assessment is a structured procedure that checks whether a medical device meets the essential safety and performance requirements of the MDR. Depending on the risk class of the product, different procedures are prescribed, ranging from self-certification (only for Class I) to comprehensive reviews by notified bodies. The result of a successful conformity assessment is the CE marking, which enables market access in the European Economic Area.
[11.08.2025]
As a Notified Body, we carry out the conformity assessment process according to a clearly structured procedure:
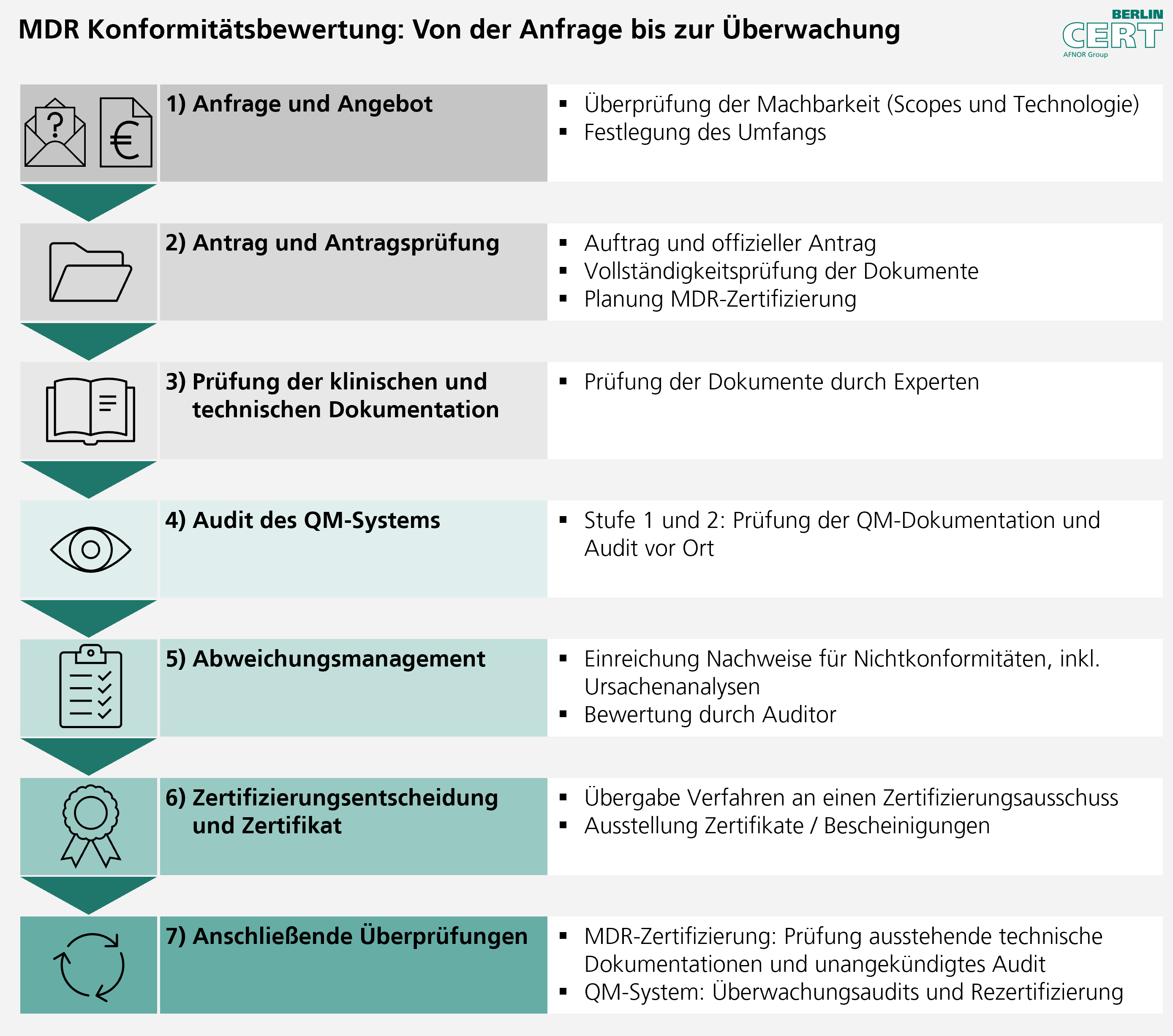
[11.08.2025]
EN ISO 13485 and the MDR are closely related, but complement each other in terms of their requirements and objectives.
Complementary requirements
- EN ISO 13485: Defines the requirements for the quality management system for medical devices
- MDR: Sets out legal requirements for access to the EU market
EN ISO 13485 certification is not a legal requirement for MDR compliance, but it is the recognised state of the art for fulfilling the QM requirements of the MDR. A quality management system certified according to EN ISO 13485 already fulfils many of the organisational and procedural elements required by the MDR.
The MDR provides the specific details of the regulatory requirements for the European Economic Area that are left open in EN ISO 13485.
Differences and Additions
The MDR goes beyond the requirements of ISO 13485 in some areas:
- More specific requirements for clinical evaluation
- More detailed regulations for post-market surveillance
- More comprehensive transparency requirements (EUDAMED)
- Stricter regulations for economic operators
[11.08.2025]
- Market access: Legal access to the European market with over 500 million potential users for your medical devices.
- Legal certainty: Proof of compliance with all legal requirements, thereby minimising liability risks.
- Competitive advantage: Differentiation through proven safety and performance in a highly regulated market.
- Trust: Strengthening the trust of patients, users and healthcare facilities in your products.
- Global recognition: MDR compliance is increasingly recognised as a quality standard outside the EU as well.
[11.08.2025]
The transition to the MDR poses significant challenges for many manufactureDocumentation and evidence:
Documentation and Evidence:
- More comprehensive technical documentation
- Higher requirements for clinical data
- Stricter evidence of biocompatibility and chemical characterisation
Process adjustments:
- Implementation of enhanced vigilance and post-market systems
- Adaptation of supply chain control
- Implementation of UDI requirements
Ressources:
- Higher time and cost expenditure for conformity assessment
- Need for specialised personnel
- Longer lead times for market launch
[11.08.2025]
The MDR introduces significant tightening and expansion:
- Stricter classification rules for medical devices
- Expanded requirements for clinical evaluations
- Introduction of Unique Device Identification (UDI)
- More comprehensive post-market surveillance
- Expansion of the scope to include products without a medical purpose
- Increased transparency through the EUDAMED database
[11.08.2025]
