Certification Process
Economic effect, among other things:
- Ensuring high quality of your own products and services
- Reducing waste and rework
- Reducing costs for warranties and follow-up services
- Elimination of incoming goods inspections
Efficiency and motivation:
- Increasing transparency regarding regulations and processes
- Active culture of error correction and improvement
Marketing:
- Certificate as proof of corporate culture and values in practice
- Differentiation from competitors and their products and services
Liability:
Evidence management in liability cases simplified through systematic handling of information and records
[11.08.2025]
The certification process is described in detail in the Berlin Cert certification regulations. In summary, it consists of the following phases:
-
Conclusion of contract
-
- Submit the information required for planning the certification process, e.g. by completing the system certification questionnaire.
- Create and confirm an offer
- Submit an application to prepare for the certification process
- Confirm the application
-
Audit Preparation
- Putting together the audit team
- Agreeing on the audit programme (procedure, dates, required documents)
-
Audit implementation stage 1 (as document review or on-site)
- Review of management system documentation
- Preparation of audit plan for stage 1, if necessary
- On-site audit, if necessary
- Summary of audit findings (stage 1 report)
- Assessment of readiness for certification
- Coordination of stage 2 audit
-
Audit implementation stage 2
- Creation of audit plan stage 2
- On-site auditing of locations in accordance with audit programme
- Introductory meeting
- Interviews with managers and employees
- Tour of the premises
- Observation of activities & conditions
- Inspection of documents
- Final meeting
- Summary of audit findings (Stage 2 report)
- If necessary, clarification of measures to remedy deviations/complaints
-
Certification Decision
- Review of audit documents by the certification body
- Independent technical review
- Certification decision made by the certification committee
-
Completion
- Signing and sending the final test report
- Issuing and sending the certificate(s)
- Obtaining and evaluating customer feedback
-
Monitoring Audits
-
Recertification
If you have any further questions about the process, please come to our free consultation hour or contact our project coordinator.
[11.08.2025]
The certification process is described in detail in the Berlin Cert certification regulations. In summary, it consists of the following phases:
- Submission of the information required for planning the certification process, e.g. by completing the system certification questionnaire.
- Preparation and confirmation of a quote
- Application for preparation for the certification process
- Confirmation of the application and introduction of the audit team
- Joint audit planning and coordination
- Preliminary assessment of QM documentation
- Certification audit at the company
- Preparation of a detailed audit report
- Implementation of corrective measures, if necessary
- Issuance of certificate and contractual agreement on use of the mark
- Surveillance audits
- Recertification audit
If you have any questions about the process, please contact the project coordinator.
[11.08.2025]
- Customer-oriented work and collegial atmosphere
- Short communication channels and direct response to your enquiries
- Competent and friendly team in a small certification body
- Many years of experience in QM certification in the medical field
- Reasonable price-performance ratio
- Wide range of certification services thanks to close cooperation with GUTcert
- Berlin Cert's integration into the Afnor Group provides additional security through an internationally represented and experienced network of experts
[11.08.2025]
ISO 13485 Certification
ISO 13485 is the international standard for quality management systems in the medical device sector. It is aimed at all organisations along the value chain – from development and manufacturing to storage and distribution of medical devices. The standard forms a central basis for the approval of medical devices in numerous markets worldwide, including Europe with the CE marking.
[11.08.2025]
The certification does not make any judgements about individual products and does not replace product certification or approval. Rather, it confirms that your management system has established the minimum requirements for processes to develop, manufacture and distribute medical devices. The quality management system according to ISO 13485 forms an excellent basis for the creation of high-quality medical devices.
[11.08.2025]
ISO 13485 certification is particularly relevant for:
Manufacturers and suppliers:
- Medical device manufacturers: From imaging procedures to implants and surgical instruments, certification is often essential for all manufacturers of medical devices.
- Suppliers: Companies that supply components for medical devices are increasingly being asked to obtain certification in order to remain in the supply chain.
Service providers and specialist suppliers:
- Medical technology service providers: Service providers for the installation, maintenance and repair of medical devices benefit from structured process design.
- Software developers in the medical field: For software as a medical device or for medical applications, ISO 13485 is a decisive proof of quality.
- Manufacturers and distributors: Organisations that assemble, sterilise, package or distribute medical devices increasingly require certification.
[11.08.2025]
- Market access: Simplified access to international markets for your medical devices, especially in Europe, the USA and Asia, where the standard is recognised as a quality benchmark.
- Legal certainty: Proof of compliance with regulatory requirements for medical devices, thereby improving your legal position vis-à-vis authorities and in the event of damage.
- Customer loyalty: Increased trust among customers, patients and healthcare facilities through demonstrable quality orientation and standardised processes.
- Process optimisation: Structured improvement of internal processes, reduction of error risks and increased efficiency throughout the entire product life cycle.
[11.08.2025]
ISO 13485 certification is based on five key pillars of quality management:
- A fully documented quality management system
- Clearly defined management responsibilities
- Systematic resource management
- Consistent control of product realisation
- Continuous measurement, analysis and improvement
- These requirements for quality management systems apply equally to medical device manufacturers, suppliers, service providers and software developers – wherever medical devices are created or processed.
[11.08.2025]
ISO 13485 differs from ISO 9001 in the following ways:
- Focus on the safety and performance of medical devices rather than general customer orientation
- Risk management as a central, consistent element in quality management
- Stricter requirements for the validation and verification of processes
- Significantly more extensive documentation requirements for medical device manufacturers
[11.08.2025]
MDR Conformity Assessment
The Medical Device Regulation (MDR, Regulation (EU) 2017/745) has been fully in force since May 2021 and replaces the previous Medical Devices Directive 93/42/EEC (MDD). As an EU regulation, it applies directly in all member states and imposes increased requirements on the safety, performance and traceability of medical devices. The MDR defines comprehensive regulations for the manufacture, distribution and monitoring of medical devices with the aim of improving patient safety.
[11.08.2025]
Conformity assessment is a structured procedure that checks whether a medical device meets the essential safety and performance requirements of the MDR. Depending on the risk class of the product, different procedures are prescribed, ranging from self-certification (only for Class I) to comprehensive reviews by notified bodies. The result of a successful conformity assessment is the CE marking, which enables market access in the European Economic Area.
[11.08.2025]
As a Notified Body, we carry out the conformity assessment process according to a clearly structured procedure:
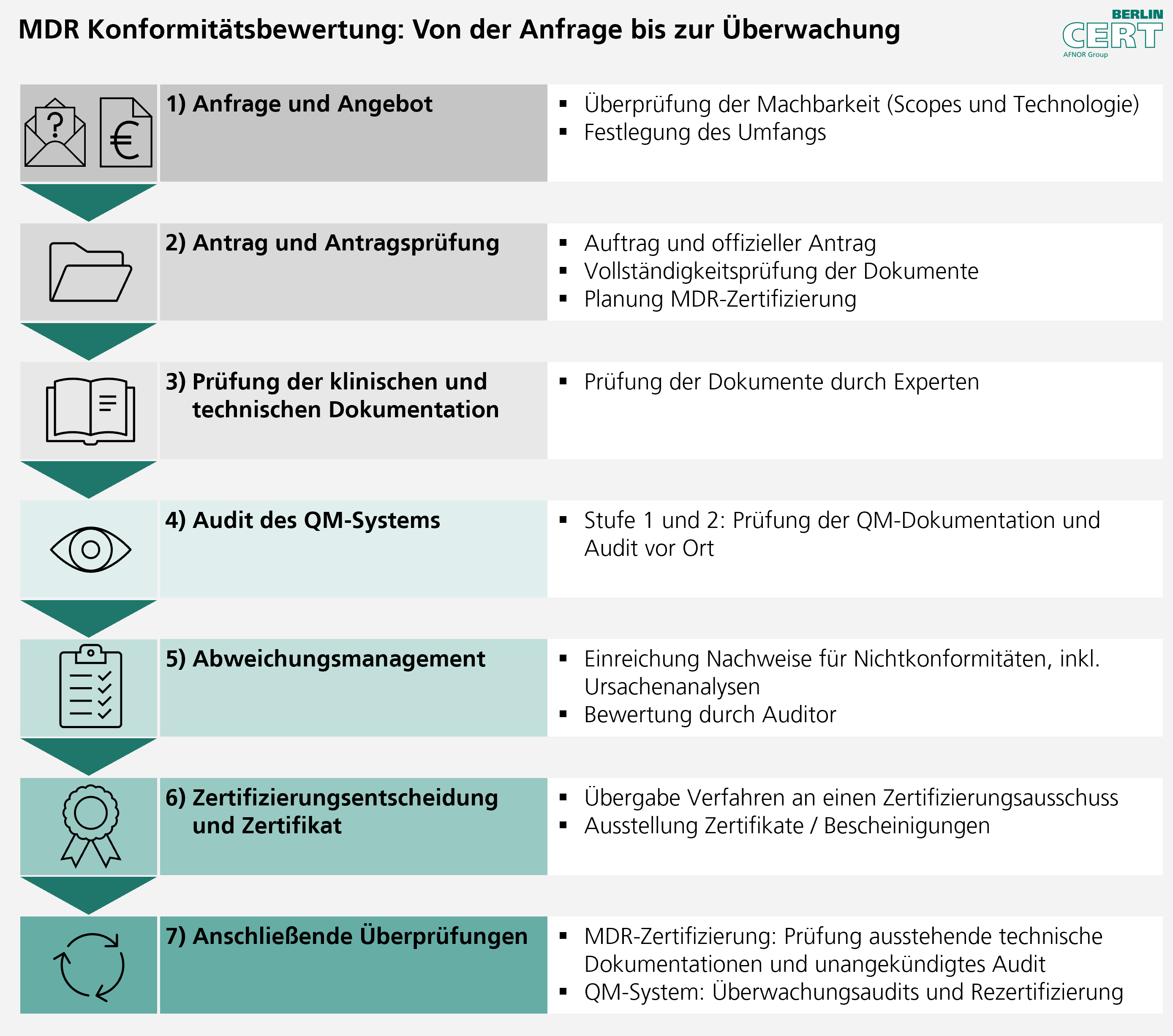
[11.08.2025]
EN ISO 13485 and the MDR are closely related, but complement each other in terms of their requirements and objectives.
Complementary requirements
- EN ISO 13485: Defines the requirements for the quality management system for medical devices
- MDR: Sets out legal requirements for access to the EU market
EN ISO 13485 certification is not a legal requirement for MDR compliance, but it is the recognised state of the art for fulfilling the QM requirements of the MDR. A quality management system certified according to EN ISO 13485 already fulfils many of the organisational and procedural elements required by the MDR.
The MDR provides the specific details of the regulatory requirements for the European Economic Area that are left open in EN ISO 13485.
Differences and Additions
The MDR goes beyond the requirements of ISO 13485 in some areas:
- More specific requirements for clinical evaluation
- More detailed regulations for post-market surveillance
- More comprehensive transparency requirements (EUDAMED)
- Stricter regulations for economic operators
[11.08.2025]
- Market access: Legal access to the European market with over 500 million potential users for your medical devices.
- Legal certainty: Proof of compliance with all legal requirements, thereby minimising liability risks.
- Competitive advantage: Differentiation through proven safety and performance in a highly regulated market.
- Trust: Strengthening the trust of patients, users and healthcare facilities in your products.
- Global recognition: MDR compliance is increasingly recognised as a quality standard outside the EU as well.
[11.08.2025]
The transition to the MDR poses significant challenges for many manufactureDocumentation and evidence:
Documentation and Evidence:
- More comprehensive technical documentation
- Higher requirements for clinical data
- Stricter evidence of biocompatibility and chemical characterisation
Process adjustments:
- Implementation of enhanced vigilance and post-market systems
- Adaptation of supply chain control
- Implementation of UDI requirements
Ressources:
- Higher time and cost expenditure for conformity assessment
- Need for specialised personnel
- Longer lead times for market launch
[11.08.2025]
The MDR introduces significant tightening and expansion:
- Stricter classification rules for medical devices
- Expanded requirements for clinical evaluations
- Introduction of Unique Device Identification (UDI)
- More comprehensive post-market surveillance
- Expansion of the scope to include products without a medical purpose
- Increased transparency through the EUDAMED database
[11.08.2025]
