Zertifizierungsverfahren
Betriebswirtschaftliche Effekt, u.a.
- Sicherstellen von hoher Qualität der eigenen Produkte und Dienstleistungen
- Reduzieren von Ausschuss und Nacharbeit
- Reduzieren der Kosten für Garantie und Folgeleistungen
- Wegfall der Wareneingangsprüfungen
Effizienz und Motiviation
- Erhöhen der Transparenz zu Regelungen und Abläufen
- Aktive Fehler- und Verbesserungskultur
Marketing
- Zertifikat als Nachweis gelebter Firmenkultur und Werte
- Abgrenzung gegenüber Wettbewerbern bzw. deren Produkten und Dienstleistungen
Haftung
- Nachweisführung im Haftungsfall durch systematische Handhabung von Informationen und Aufzeichnungen vereinfacht
[25.07.2025]
Der Ablauf der Zertifizierung ist ausführlich in der Zertifizierungsordnung der Berlin Cert beschrieben. Zusammengefasst besteht er aus den folgenden Phasen:
-
Vertragsschluss
-
- Einreichen der für die Planung des Zertifizierungsverfahrens notwendigen Informationen, z. B. durch Ausfüllen des Fragebogens Systemzertifizierung.
- Erstellen und Bestätigung eines Angebotes
- Antrag zur Vorbereitung auf das Zertifizierungsverfahren stellen
- Bestätigen des Antrags
-
Auditvorbereitung
- Zusammenstellung des Auditteams
- Abstimmung Auditprogramm (Ablauf, Termine, benötigte Unterlagen)
-
Auditdurchführung Stufe 1 (als Dokumentenprüfung oder vor Ort)
- Prüfung der Managementsystem-Dokumentation
- ggf. Erstellung des Auditplans für Stufe 1
- ggf. vor Ort Auditierung
- Zusammenfassung der Auditfeststellungen (Stufe 1-Bericht)
- Bewertung der Zertifizierungsreife
- Abstimmung zum Stufe 2-Audit
-
Auditdurchführung Stufe 2
- Erstellung Auditplan Stufe 2
- Vor-Ort Auditierung der Standorte gemäß Auditprogramm
- Einführungsgespräch
- Interviews mit Führungskräften und Mitarbeitern
- Betriebsrundgang
- Beobachtung von Tätigkeiten & Zuständen
- Dokumenteneinsicht
- Abschlussgespräch
- Zusammenfassung der Auditfeststellungen (Stufe 2-Bericht)
- Ggf. Klärung der Maßnahmen zur Behebung von Abweichungen / Beanstandungen
-
Zertifizierungsentscheidung
- Prüfung der Auditunterlagen durch die Zertifizierungsstelle
- unabhängige fachliche Überprüfung
- Treffen der Zertifizierungsentscheidung durch Zertifizierungsausschuss
-
Abschluss
- Signatur und Versand des finalen Prüfberichts
- Ausstellung und Versand des/r Zertifikate(s)
- Kundenfeedback einholen und auswerten
-
Überwachungsaudits
-
Rezertifizierungs
Bei weiteren Fragen zu dem Ablauf: kommen Sie in unsere kostenlose Sprechstunde, oder sprechen Sie unsere Projektkoordination an.
[25.07.2025]
- Kundenorientierte Arbeit und kollegiale Atmosphäre
- Kurze Wege und direkte Reaktion auf Ihre Anfragen
- Kompetentes und freundliches Team in einer kleinen Zertifizierungsstelle
- Langjährige Erfahrung in der QM-Zertifizierung im medizinischen Umfeld
- Angemessenes Preis-Leistungs-Verhältnis
- großes Leistungsspektrum an Zertifizierungen durch enge Zusammenarbeit mit der GUTcert
- Die Einbettung der Berlin Cert in die Afnor Group gibt zusätzliche Sicherheit eines international vertretenen und erfahrenen Expertennetzwerkes
[22.07.2025]
ISO 13485 Zertifizierung
Die ISO 13485 ist der internationale Standard für Qualitätsmanagementsysteme im Medizinproduktesektor. Sie richtet sich an alle Organisationen entlang der Wertschöpfungskette – von der Entwicklung über die Herstellung bis hin zu Lagerung und Vertrieb von Medizinprodukten. Die Norm bildet eine zentrale Grundlage für die Zulassung von Medizinprodukten in zahlreichen Märkten weltweit, darunter Europa mit der CE-Kennzeichnung.
[14.07.2025]
Die Zertifizierung gibt keine Urteile über einzelne Produkte ab und ersetzt keine Produktzertifizierung oder -zulassung. Sie bestätigt vielmehr, dass Ihr Managementsystem die Mindestanforderungen an Prozesse etabliert hat, um Medizinprodukte zu entwickeln, herzustellen und zu vertreiben. Das Qualitätsmanagementsystem nach ISO 13485 bildet eine exzellente Grundlage, mit der qualitativ hochwertige Medizinprodukte entstehen können.
[31.07.2025]
Die ISO 13485 Zertifizierung ist besonders relevant für:
Hersteller und Zulieferer:
- Medizinproduktehersteller: Von bildgebenden Verfahren über Implantate bis zu chirurgischen Instrumenten – für alle Hersteller von Medizinprodukten ist die Zertifizierung oft unerlässlich.
- Zulieferer: Unternehmen, die Komponenten für Medizinprodukte liefern, werden zunehmend zur Zertifizierung aufgefordert, um in der Lieferkette bestehen zu können.
Dienstleister und Spezialanbieter:
- Medizintechnik-Dienstleister: Serviceanbieter für Installation, Wartung und Reparatur medizintechnischer Geräte profitieren von der strukturierten Prozessgestaltung.
- Softwareentwickler im Medizinbereich: Bei Software als Medizinprodukt oder für medizinische Anwendungen ist die ISO 13485 ein entscheidender Qualitätsnachweis.
- Fertiger und Händler: Organisationen, die Medizinprodukte zusammenbauen, sterilisieren, verpacken oder vertreiben, benötigen zunehmend eine Zertifizierung.
[14.07.2025]
- Marktzugang: Vereinfachte Erschließung internationaler Märkte für Ihre Medizinprodukte, insbesondere in Europa, USA und Asien, wo die Norm als Qualitätsstandard anerkannt ist.
- Rechtssicherheit: Nachweis der Erfüllung regulatorischer Anforderungen an Medizinprodukte und damit verbesserte juristische Position gegenüber Behörden und im Schadensfall.
- Kundenbindung: Steigerung des Vertrauens von Kunden, Patienten und Gesundheitseinrichtungen durch nachweisbare Qualitätsorientierung und standardisierte Prozesse.
- Prozessoptimierung: Strukturierte Verbesserung interner Abläufe, Reduzierung von Fehlerrisiken und Effizienzsteigerung im gesamten Produktlebenszyklus.
[14.07.2025]
Die ISO 13485 Zertifizierung basiert auf fünf wesentlichen Säulen des Qualitätsmanagements:
- Ein vollständig dokumentiertes Qualitätsmanagementsystem
- Klar definierte Management-Verantwortlichkeiten
- Systematisches Ressourcenmanagement
- Durchgängige Kontrolle der Produktrealisierung
- Kontinuierliche Messung, Analyse und Verbesserung
- Diese Anforderungen an Qualitätsmanagementsysteme gelten für Medizingerätehersteller, Zulieferer, Dienstleister und Softwareentwicklergleichermaßen – überall dort, wo Medizinprodukte entstehen oder bearbeitet werden.
[14.07.2025]
MDR Konformitätsbewertung
Die Medical Device Regulation (MDR, Verordnung (EU) 2017/745) ist seit Mai 2021 vollständig in Kraft und ersetzt die bisherige Medizinprodukterichtlinie 93/42/EWG (MDD). Als EU-Verordnung gilt sie unmittelbar in allen Mitgliedstaaten und stellt erhöhte Anforderungen an die Sicherheit, Leistung und Rückverfolgbarkeit von Medizinprodukten. Die MDR definiert umfassende Regelungen für die Herstellung, den Vertrieb und die Überwachung von Medizinprodukten mit dem Ziel, den Patientenschutz zu verbessern.
[14.07.2025]
Die Konformitätsbewertung ist ein strukturiertes Verfahren, bei dem geprüft wird, ob ein Medizinprodukt die grundlegenden Sicherheits- und Leistungsanforderungen der MDR erfüllt. Je nach Risikoklasse des Produkts sind unterschiedliche Verfahren vorgeschrieben, die von der Selbstzertifizierung (nur bei Klasse I) bis hin zu umfassenden Überprüfungen durch Benannte Stellen reichen. Das Ergebnis einer erfolgreichen Konformitätsbewertung ist die CE-Kennzeichnung, die den Marktzugang im Europäischen Wirtschaftsraum ermöglicht.
[14.07.2025]
Als Benannte Stelle führen wir den Konformitätsbewertungsprozess nach einem klar strukturierten Verfahren durch:
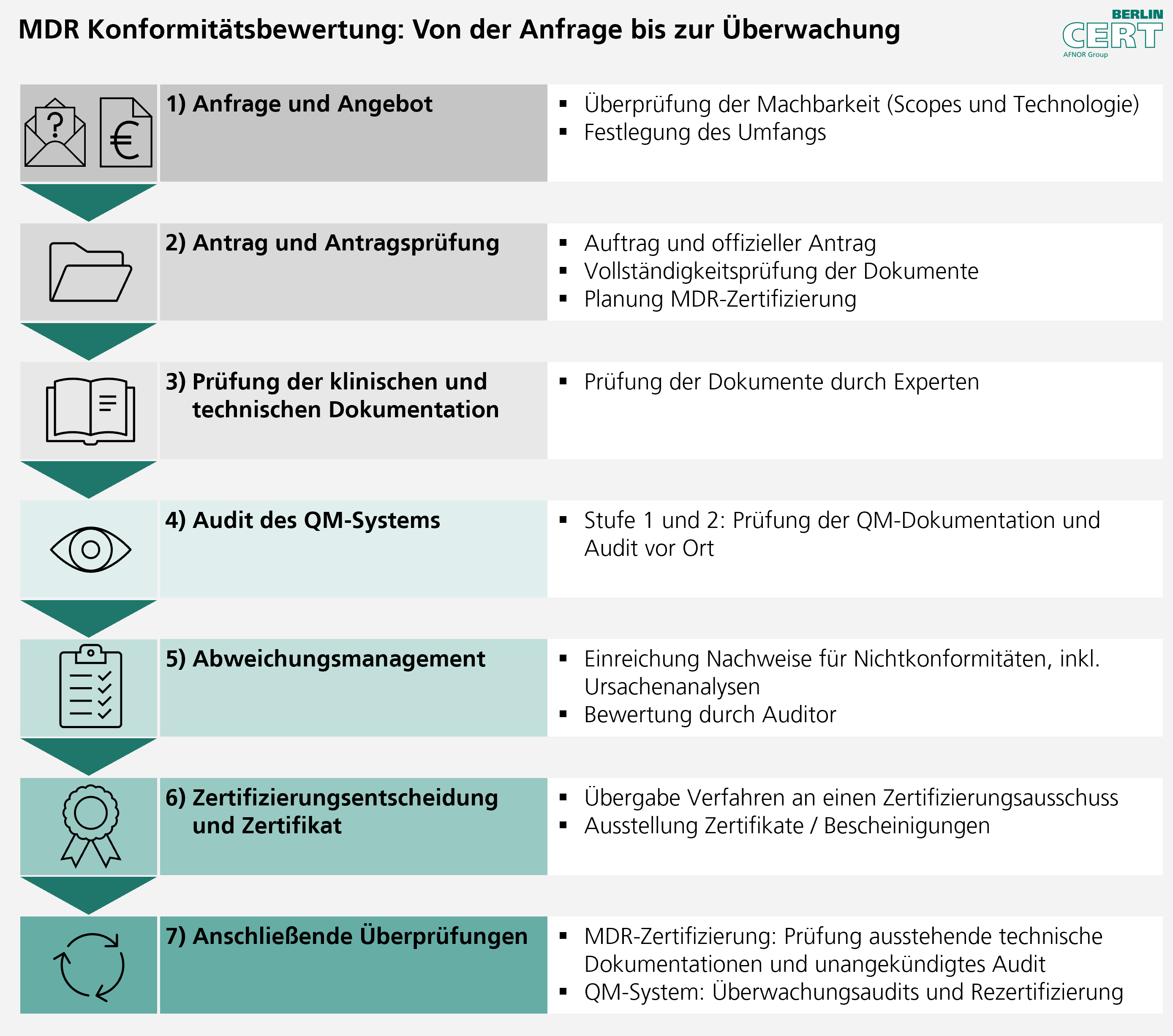
[14.07.2025]
Die EN ISO 13485 und die MDR stehen in einem engen Zusammenhang, ergänzen sich jedoch in ihren Anforderungen und Zielsetzungen.
Komplementäre Anforderungen
- EN ISO 13485: Definiert die Anforderungen an das Qualitätsmanagementsystem für Medizinprodukte
- MDR: Legt gesetzliche Anforderungen für den EU-Marktzugang fest
Die EN ISO 13485-Zertifizierung ist keine rechtliche Voraussetzung für die MDR-Konformität, bildet jedoch den anerkannten Stand der Technik für die Erfüllung der QM-Anforderungen der MDR. Ein nach EN ISO 13485 zertifiziertes Qualitätsmanagementsystem erfüllt bereits viele der in der MDR geforderten organisatorischen und prozessualen Elemente
Die MDR liefert die spezifische Ausgestaltung der in der EN ISO 13485 offen gehaltenen regulatorischen Anforderungen für den europäischen Wirtschaftsraum.
Unterschiede und Ergänzungen
Die MDR geht in einigen Bereichen über die Anforderungen der ISO 13485 hinaus:
- Spezifischere Anforderungen an die klinische Bewertung
- Detailliertere Regelungen zur Überwachung nach der Inverkehrbringung
- Umfassendere Transparenzanforderungen (EUDAMED)
- Strengere Regelungen für wirtschaftliche Akteure
[14.07.2025]
- Marktzugang: Legaler Zugang zum europäischen Markt mit über 500 Millionen potenziellen Nutzern für Ihre Medizinprodukte.
- Rechtssicherheit: Nachweis der Erfüllung aller gesetzlichen Anforderungen und damit Minimierung von Haftungsrisiken.
- Wettbewerbsvorteil: Differenzierung durch nachgewiesene Sicherheit und Leistungsfähigkeit in einem stark regulierten Markt.
- Vertrauen: Stärkung des Vertrauens von Patienten, Anwendern und Gesundheitseinrichtungen in Ihre Produkte.
- Globale Anerkennung: Die MDR-Konformität wird zunehmend als Qualitätsstandard auch außerhalb der EU anerkannt.
[14.07.2025]
Die Umstellung auf die MDR stellt viele Hersteller vor erhebliche Herausforderungen:
Dokumentation und Nachweise:
- Umfangreichere technische Dokumentation
- Höhere Anforderungen an klinische Daten
- Strengere Nachweise zur Biokompatibilität und chemischen Charakterisierung
Prozessanpassungen:
- Implementierung erweiterter Vigilanz- und Nachmarktsysteme
- Anpassung der Lieferkettenkontrolle
- Umsetzung der UDI-Anforderungen
Ressourcen:
- Höherer Zeit- und Kostenaufwand für die Konformitätsbewertung
- Bedarf an spezialisierten Fachkräften
- Längere Vorlaufzeiten für die Markteinführung
[14.07.2025]
